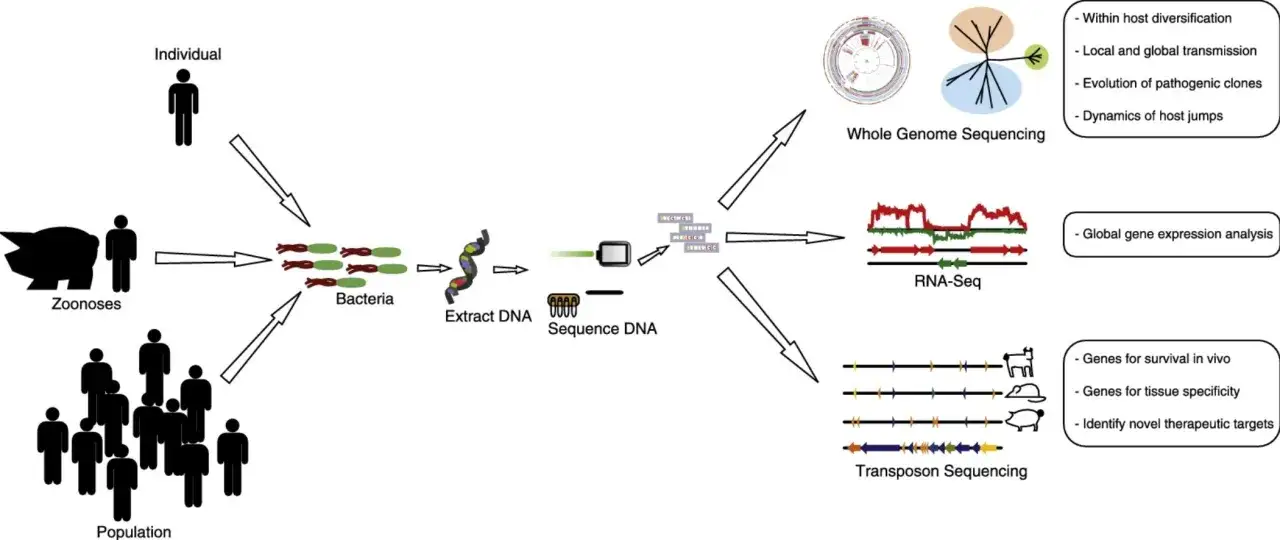
Le séquençage haut débit a changé la manière dont on lit l’ADN et l’ARN: on ne regarde plus un gène isolé, on analyse des millions de fragments en parallèle. Pour la biologie et l’évolution, cela ouvre des questions beaucoup plus larges, de la parenté entre espèces aux mécanismes d’adaptation, en passant par les populations anciennes et les génomes difficiles à assembler. Je vais aller droit au but: principe, déroulé d’une analyse, usages vraiment utiles, limites à connaître et critères concrets pour choisir la bonne approche.
Ce qu’il faut retenir avant de lire la suite
- La technologie lit des fragments en masse, puis un logiciel les aligne ou les assemble pour reconstituer une séquence exploitable.
- Une lecture courte tourne souvent autour de 300 à 400 paires de bases, alors que les lectures longues couvrent plusieurs milliers de bases, parfois bien davantage.
- En biologie évolutive, elle sert surtout à reconstruire des parentés, suivre la diversité génétique, détecter des signatures de sélection et travailler sur l’ADN ancien.
- Le choix entre lectures courtes, longues ou ciblées dépend moins de la mode que de la question posée et de l’état de l’échantillon.
- Les erreurs les plus fréquentes viennent d’une couverture insuffisante, d’une contamination, d’un biais de référence ou d’une analyse bioinformatique trop vite validée.
Ce que mesure vraiment un séquenceur
Je commence toujours par une précision utile: un séquenceur ne “voit” pas un génome entier d’un seul bloc. Il lit des fragments, appelés lectures ou reads, puis un logiciel reconstruit l’ensemble à partir de ces pièces. C’est ce passage du fragment au signal biologique qui fait toute la puissance de la méthode, mais aussi ses limites.
| Terme | Ce que cela signifie | Pourquoi c’est important |
|---|---|---|
| Lecture | Séquence courte produite par l’instrument | Plus elle est longue et propre, plus l’analyse est simple |
| Couverture | Nombre de fois qu’une base est lue | Elle conditionne la fiabilité des variantes détectées |
| Alignement | Placement des lectures sur un génome de référence | Utile quand une référence existe et que l’on compare des échantillons |
| Assemblage de novo | Reconstruction sans référence préalable | Essentiel pour les espèces non modèles ou les génomes très remaniés |
| Variant | Différence par rapport à une séquence de référence | Permet de repérer mutations, polymorphismes et signatures évolutives |
Dans les usages courants, les lectures courtes sont souvent autour de 300 à 400 paires de bases, ce qui suffit très bien pour beaucoup de comparaisons, mais devient moins confortable dès qu’il faut traverser des régions répétées ou reconstruire de grands réarrangements. Une fois cette base posée, la vraie question devient le chemin qui mène de l’échantillon brut à une interprétation biologique solide.
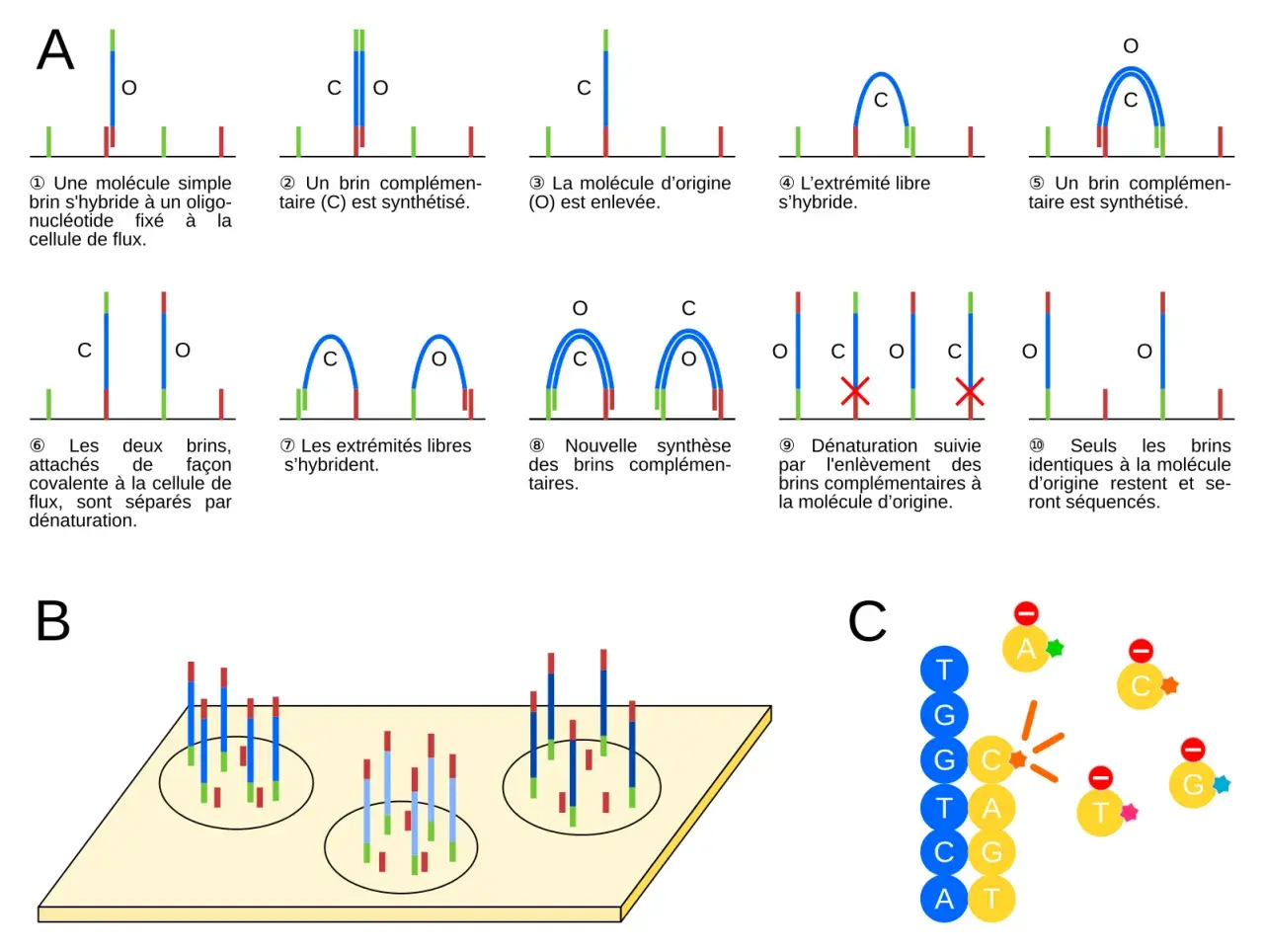
Comment un échantillon devient une donnée interprétable
Le flux de travail est plus simple à comprendre qu’il n’y paraît, à condition de ne pas le réduire à “on met de l’ADN dans la machine”. En pratique, chaque étape peut introduire un biais, et c’est souvent là que se joue la qualité finale du résultat.
- Extraction de l’ADN ou de l’ARN : la qualité du matériel de départ compte énormément, surtout pour les échantillons anciens, dégradés ou très riches en contaminants.
- Fragmentation et préparation de bibliothèque : les fragments sont préparés avec des adaptateurs, des petites séquences qui permettent de les lire sur la plateforme choisie.
- Séquençage : l’instrument lit les fragments en parallèle, parfois des millions en une seule série d’opérations.
- Appel de bases : le signal brut est converti en lettres A, C, G et T, avec une estimation de qualité pour chaque position.
- Contrôle qualité : on retire les lectures trop courtes, trop bruitées ou contaminées par des adaptateurs mal coupés.
- Alignement ou assemblage : les lectures sont soit alignées sur une référence, soit assemblées sans référence si le contexte l’exige.
- Analyse biologique : on cherche des variantes, des gènes, des signatures de sélection, des profils d’expression ou des relations de parenté.
Deux notions reviennent tout le temps dans les articles sérieux: la couverture et la qualité. Une couverture élevée réduit les faux positifs et permet d’oser des conclusions plus fines; une qualité médiocre, elle, donne l’illusion d’un résultat riche alors que le signal est fragile. C’est cette étape de traitement qui transforme une masse de fragments en information scientifique utilisable, ce qui nous amène aux usages concrets en biologie et en évolution.
Pourquoi cette approche a bouleversé la biologie de l’évolution
Le point décisif, à mes yeux, n’est pas seulement la vitesse ou le volume. C’est le fait qu’on puisse enfin comparer des génomes à grande échelle, sur des espèces, des populations ou des individus, sans rester limité à quelques marqueurs. En évolution, cela change la profondeur de l’enquête.
Reconstituer des parentés plus finement
Les arbres phylogénétiques se construisaient déjà avant le séquençage à haut débit, mais la méthode a fait passer la discipline d’une poignée de gènes à des milliers de loci. Résultat: on distingue mieux les lignées proches, on repère les convergences trompeuses et on évite de tirer des conclusions trop rapides à partir d’un seul marqueur.
Suivre l’évolution des populations
Quand je veux comprendre une population, je m’intéresse aux fréquences des variants, à leur répartition géographique et à la façon dont elles changent dans le temps. Cette approche permet de repérer une sélection locale, une dérive génétique forte, un goulot d’étranglement ou un flux de gènes entre populations. C’est particulièrement utile pour étudier des espèces qui se sont adaptées à un milieu précis, par exemple un changement de température, de salinité ou d’altitude.
Lire l’ADN ancien et les génomes difficiles
Les échantillons anciens posent un problème très concret: l’ADN est fragmenté, abîmé et parfois contaminé. C’est précisément là que le séquençage à haut débit a apporté une méthode de travail robuste, parce qu’il tolère des fragments très courts et produit assez de profondeur pour faire émerger un signal exploitable. En paléogénomique, cela permet d’éclairer des migrations, des hybridations ou des épisodes d’extinction avec une précision autrefois inaccessible.
Lire aussi : Cycle Cellulaire - Comprendre ses étapes et contrôles
Comparer le vivant au niveau moléculaire
Pour les organismes non modèles, l’intérêt est encore plus net. On n’a pas toujours un génome de référence de grande qualité, mais on peut quand même séquencer, assembler et comparer. Cela ouvre des études sur des insectes, des plantes, des champignons, des bactéries ou des communautés microbiennes qui étaient longtemps sous-étudiés faute d’outils adaptés.
Autrement dit, la méthode n’est pas seulement un outil de lecture, c’est un instrument de comparaison à grande échelle. La suite logique consiste donc à choisir la bonne stratégie selon la question biologique, pas selon la seule puissance technique disponible.
Choisir la bonne stratégie selon la question biologique
Je vois souvent la même erreur: vouloir “le plus de données possible” au lieu de choisir le type de données qui répond vraiment à la question. Or, en biologie évolutive, la meilleure stratégie n’est pas toujours la plus ambitieuse; c’est celle qui équilibre résolution, coût, qualité des échantillons et temps d’analyse.
| Approche | Ce qu’elle répond | Atout principal | Limite principale | Cas d’usage en évolution |
|---|---|---|---|---|
| Lectures courtes | Variantes ponctuelles, comparaison fine entre échantillons | Rapide, très robuste, bon rapport coût/données | Moins bonne lecture des régions répétées et des grands réarrangements | Population genetics, phylogénies sur génomes proches, ADN ancien court |
| Lectures longues | Assemblage complet, variants structurels, régions complexes | Traverse mieux les répétitions et améliore l’assemblage | Demande souvent une meilleure qualité d’ADN et un budget plus élevé | Génomes non modèles, hybridation, réarrangements, génomes très fragmentés à reconstruire |
| Séquençage ciblé | Quelques gènes, régions codantes, marqueurs choisis | Très efficace sur beaucoup d’échantillons | Vue plus étroite du génome | Phylogénie appliquée, suivi d’aires de répartition, panels de variants |
| Transcriptome | Gènes exprimés dans une condition donnée | Montre ce qui s’active ou se tait | Dépend du tissu, du stade et du contexte biologique | Adaptation au stress, évolution de la régulation génétique |
| Métagénomique | Communautés d’organismes présents dans un échantillon | Évite la culture préalable | Analyse complexe, mélange de génomes et biais d’abondance | Évolution des microbiotes, écologie microbienne, symbioses |
Si je devais résumer en une phrase: lectures courtes pour la précision statistique, lectures longues pour la structure et l’assemblage. Pour l’ADN dégradé, les lectures courtes restent souvent plus réalistes; pour les génomes répétitifs ou les remaniements chromosomiques, les longues prennent l’avantage. Et même dans les projets bien conçus, plusieurs pièges peuvent encore déformer l’histoire que l’on croit lire.
Les pièges qui faussent l’interprétation
Le séquençage à haut débit produit des données puissantes, mais pas magiques. Les erreurs les plus coûteuses ne viennent pas toujours de la machine; elles viennent souvent d’un mauvais cadrage du projet ou d’une analyse trop confiante.
- Une couverture insuffisante : elle laisse passer des variants réels et fait grimper le bruit de fond.
- La contamination : particulièrement critique pour l’ADN ancien, les échantillons environnementaux ou les faibles quantités d’ADN.
- Le biais de référence : si le génome de référence est trop éloigné, on sous-estime certaines variantes ou on les aligne mal.
- Les régions répétées : elles provoquent des assemblages ambiguës, surtout avec des lectures courtes.
- Les duplicats PCR : ils donnent l’illusion d’une profondeur plus forte qu’elle ne l’est réellement.
- Les paramètres bioinformatiques : une valeur de filtrage trop stricte fait disparaître du vrai signal; trop laxiste, elle laisse entrer des artefacts.
Je conseille aussi de ne jamais interpréter une variante isolée comme une preuve d’adaptation sans contexte: il faut regarder la répétabilité, la structure de population, le type de mutation, et si possible la validation par une autre méthode. Dans la pratique, une bonne conclusion évolutive repose autant sur le design de l’étude que sur la qualité du séquençage lui-même.
Les vérifications qui évitent de surinterpréter l’histoire du vivant
Avant de publier une conclusion sur une espèce, une population ou une adaptation, je vérifie toujours quelques points très concrets. Ce sont des garde-fous simples, mais ils font souvent la différence entre une lecture solide et une lecture séduisante mais fragile.
- La question est-elle assez précise ? Comparer des espèces, suivre une migration ou détecter une sélection ne demande pas le même type de données.
- L’échantillon est-il adapté ? Un ADN très dégradé oriente vers des lectures courtes et une stratégie de capture plus conservatrice.
- La profondeur est-elle cohérente ? Un jeu de données trop mince peut suffire pour un criblage, mais pas pour une interprétation fine.
- Les contrôles existent-ils ? Témoin négatif, réplicat, contrôle de contamination et validation orthogonale sont des réflexes sains.
- Le pipeline est-il reproductible ? Les paramètres d’alignement, de filtrage et d’appel de variants doivent être documentés.
- Les métadonnées sont-elles complètes ? Origine, date, tissu, environnement et méthode de prélèvement comptent presque autant que la séquence.
Quand ces points sont verrouillés, le séquençage à haut débit devient un outil d’une précision remarquable pour comprendre la diversité du vivant. Ce n’est pas seulement une technologie de lecture; c’est une méthode pour relier des fragments d’information à une histoire évolutive cohérente, à condition de laisser les données parler avec prudence plutôt qu’avec excès de confiance.