Les repères essentiels à garder en tête
- La forme adulte de la maladie dure souvent 15 à 20 ans après le début des symptômes, avec des fourchettes plus larges dans certaines synthèses cliniques.
- La forme juvénile évolue en général plus vite, avec une survie souvent située autour de 10 à 15 ans après l’apparition des signes.
- Le chiffre utile n’est pas la date du diagnostic, mais bien le moment où les symptômes commencent vraiment à s’installer.
- La déglutition, la nutrition, les chutes et les infections respiratoires comptent parmi les facteurs qui pèsent le plus sur l’évolution.
- Il n’existe pas encore de traitement curatif, mais la prise en charge multidisciplinaire améliore nettement la qualité de vie.
- Un test génétique peut confirmer le risque, mais il ne permet pas de prédire une date précise d’apparition ni une durée de vie exacte.
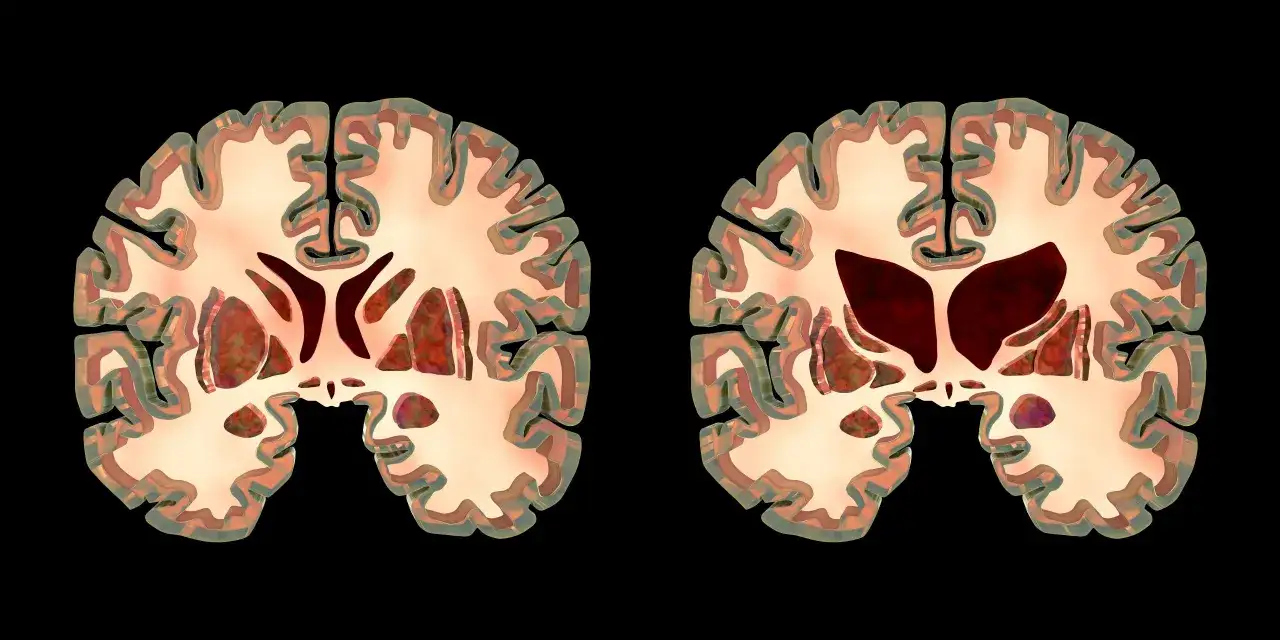
Quelle est l’espérance de vie moyenne dans la maladie de Huntington
Je préfère parler de fourchette plutôt que de moyenne unique, parce que la maladie évolue de façon très différente selon l’âge de début, la vitesse de progression et les complications associées. Pour la forme la plus fréquente, celle de l’adulte, on retient souvent une survie d’environ 15 à 20 ans après l’apparition des premiers symptômes. Certaines synthèses médicales françaises donnent une plage plus large, autour de 20 à 30 ans, ce qui ne contredit pas la première estimation : cela reflète surtout des populations étudiées différentes et des débuts de maladie pas toujours définis de la même manière.
| Forme de la maladie | Âge de début habituel | Durée de vie après les premiers symptômes | Ce qu’il faut retenir |
|---|---|---|---|
| Forme adulte | Le plus souvent entre 30 et 50 ans | Environ 15 à 20 ans, parfois davantage | C’est la forme la plus fréquente et la plus étudiée |
| Forme juvénile | Avant 20 ans, parfois durant l’enfance | Souvent 10 à 15 ans | L’évolution est généralement plus rapide |
Le point clé, c’est que ce chiffre concerne le début des symptômes, pas la date du diagnostic ni celle du test génétique. Entre les deux, il peut se passer du temps, et ce décalage fausse souvent la lecture du pronostic. C’est justement pour cela qu’il faut regarder de près les facteurs qui font varier l’évolution.
Pourquoi les chiffres varient autant d’une personne à l’autre
La maladie de Huntington n’avance pas au même rythme chez tout le monde. Deux personnes de la même famille peuvent partager la mutation, mais vivre des trajectoires très différentes. À mon sens, c’est l’un des points les plus difficiles à comprendre pour les proches : le gène explique le risque, mais il ne dessine pas un calendrier précis.
L’âge du début change tout
Plus les symptômes apparaissent tôt, plus la période d’évolution est souvent courte. Une personne chez qui la maladie se déclare à 35 ans n’a pas le même parcours qu’une autre chez qui elle débute après 55 ans. Ce n’est pas seulement une question de nombre d’années restantes, mais aussi de rythme de perte d’autonomie, de maintien de la marche, de la parole et de la déglutition.
Le profil génétique n’est pas qu’un détail technique
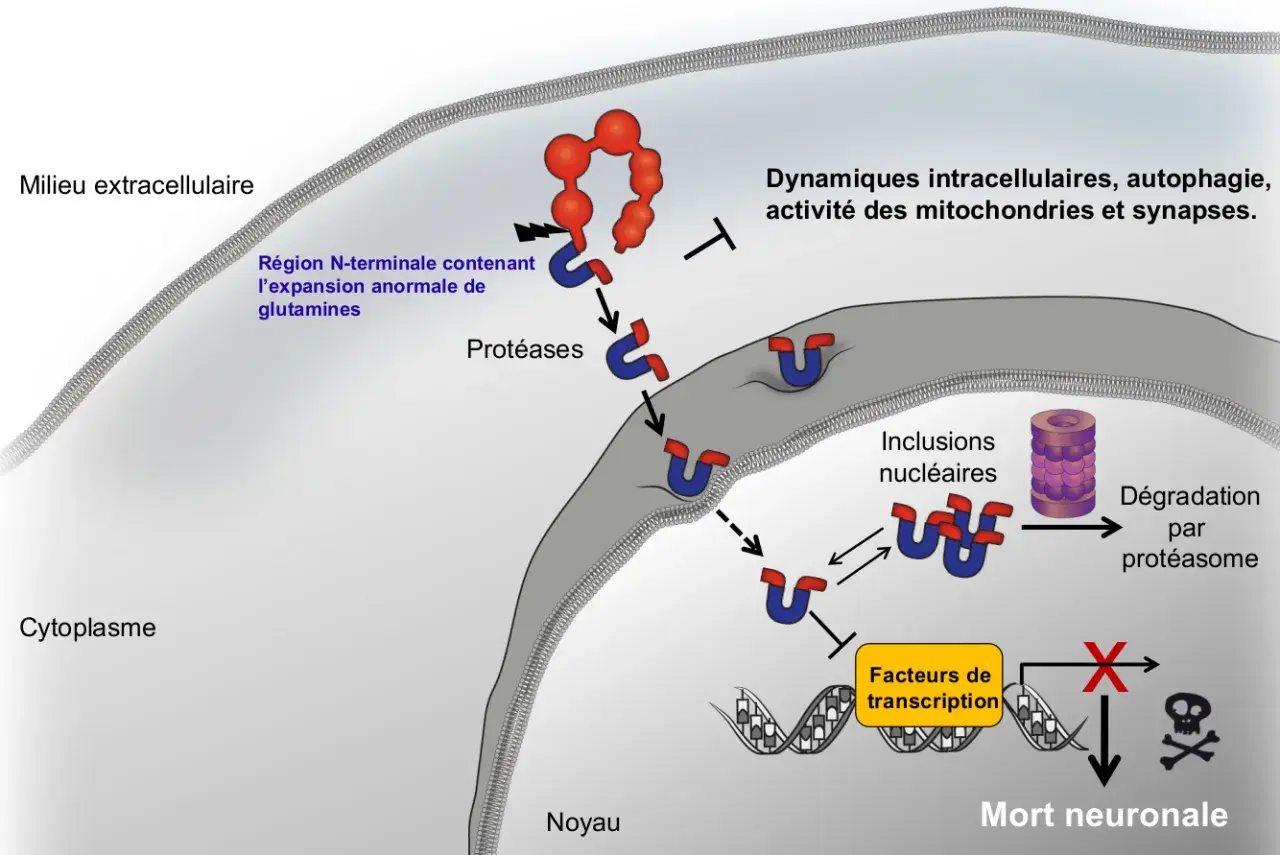
La mutation concerne le gène HTT, qui porte une répétition de triplets CAG. Concrètement, plus cette répétition est longue, plus l’apparition de la maladie est souvent précoce. C’est une donnée utile pour comprendre le risque familial, mais elle ne suffit toujours pas à prédire l’évolution exacte d’un individu.
La prise en charge modifie la trajectoire fonctionnelle
La progression biologique ne disparaît pas, mais une prise en charge bien structurée peut limiter certaines complications. Je pense notamment à l’orthophonie, au travail sur l’alimentation, à la kinésithérapie et à l’accompagnement psychiatrique. Ces soins ne changent pas tout, mais ils évitent souvent que des problèmes secondaires accélèrent inutilement le déclin.
Autrement dit, il existe une maladie de base, mais aussi une manière de la vivre et de la surveiller, et cette seconde dimension compte plus qu’on ne l’imagine. C’est ce que montrent très concrètement les complications les plus fréquentes.
Les complications qui influencent le plus le pronostic
Dans la maladie de Huntington, le pronostic dépend rarement d’un seul symptôme. Ce sont plutôt plusieurs fragilités qui s’additionnent : mouvements involontaires, troubles cognitifs, difficultés de communication, puis problèmes de déglutition et de nutrition. Les complications respiratoires finissent souvent par peser lourd.
- La déglutition devient difficile, ce qui augmente le risque de fausse route et de toux pendant les repas.
- La perte de poids peut s’installer, parfois même quand l’appétit semble encore présent.
- Les infections respiratoires, en particulier quand les fausses routes se répètent, fragilisent l’état général.
- Les chutes et les traumatismes augmentent quand la marche devient instable ou que les mouvements sont trop amples.
- Les troubles psychiatriques, comme la dépression, l’irritabilité ou l’anxiété, compliquent l’adhésion aux soins et la vie de famille.
Le détail important, c’est que la maladie ne se résume pas à des mouvements involontaires. Quand la parole, l’alimentation et la sécurité au quotidien commencent à être touchées, c’est souvent là que le parcours s’accélère. D’où l’intérêt d’agir tôt sur les soins de support.
Ce que les traitements peuvent réellement apporter
Il n’existe pas encore de traitement capable de guérir la maladie ou d’en arrêter l’évolution. En revanche, on peut soulager certains symptômes, réduire les complications et préserver l’autonomie aussi longtemps que possible. C’est une nuance essentielle, parce qu’elle évite deux pièges : attendre un remède miracle ou, au contraire, croire qu’il n’y a rien à faire.
La stratégie la plus utile repose généralement sur plusieurs axes complémentaires :
- Les médicaments peuvent aider à contrôler certains mouvements involontaires et certains symptômes psychiatriques.
- L’orthophonie sert à sécuriser la parole et surtout la déglutition quand les repas deviennent plus compliqués.
- La kinésithérapie et l’ergothérapie aident à garder la mobilité, à prévenir les chutes et à adapter le domicile.
- L’accompagnement nutritionnel devient crucial si le poids baisse ou si manger fatigue trop.
- Le suivi psychologique ou psychiatrique est souvent indispensable, pour le patient comme pour les proches.
Je retiens surtout une chose : plus la prise en charge est coordonnée, plus on évite que des symptômes secondaires prennent le dessus. C’est exactement là que la logique de soin change, et cela mène naturellement à la question du test génétique et de la famille.
Ce que le test génétique dit, et ce qu’il ne dit pas
La maladie de Huntington se transmet sur un mode autosomique dominant, ce qui signifie qu’une personne porteuse de la mutation peut la transmettre à chacun de ses enfants avec un risque de 50 %. Ce chiffre est simple à comprendre, mais il ne dit pas tout : il ne permet ni de prédire l’âge d’apparition, ni de savoir si les premiers signes seront moteurs, cognitifs ou psychiatriques.
Un résultat positif ne donne pas une date
Un test génétique positif confirme le risque de développer la maladie, mais il ne fixe pas une échéance précise. C’est souvent un malentendu chez les familles : on voudrait transformer un résultat biologique en calendrier, alors que la réalité est bien plus nuancée. L’âge d’apparition, la vitesse de progression et la durée de vie restent très variables.
Le conseil génétique est indispensable
Avant un test prédictif, l’encadrement par une équipe spécialisée est vraiment nécessaire. Le résultat ne touche pas seulement la personne testée : il a des conséquences sur les enfants, les frères et sœurs, la vie professionnelle, les projets parentaux et parfois l’équilibre psychologique de toute la famille. À mon sens, c’est une étape qu’il ne faut jamais banaliser.
Lire aussi : L'oreille - Comment ça marche et comment la protéger ?
Les proches ont aussi besoin d’informations claires
Quand un cas de Huntington touche une famille, l’objectif n’est pas seulement de poser un nom sur la maladie. Il faut aussi aider chacun à comprendre ce que signifie le risque, quand consulter, quels signes surveiller et comment organiser le suivi si les symptômes débutent. Cette clarification évite beaucoup d’angoisse inutile et quelques décisions hâtives.
Une fois ces repères posés, la question la plus utile n’est plus seulement “combien de temps”, mais “comment accompagner au mieux la personne concernée”. C’est là que la lecture du pronostic devient vraiment concrète.
Les repères à garder pour lire ce pronostic avec justesse
Si je devais résumer la situation en une phrase, je dirais ceci : la maladie de Huntington réduit en moyenne l’espérance de vie, mais la variation individuelle reste importante et la qualité de la prise en charge compte énormément. Le meilleur réflexe consiste donc à raisonner en étapes de suivi, pas en date fatale.
- Surveillez de près les difficultés à avaler, la toux pendant les repas et les fausses routes.
- Faites réévaluer rapidement une perte de poids ou une fatigue qui s’installe malgré une alimentation apparemment correcte.
- Ne minimisez pas les chutes, l’isolement, la dépression ou le repli, car ces signes modifient souvent la trajectoire clinique.
En pratique, la bonne information n’est pas de savoir si la durée sera exactement de 17, 22 ou 28 ans. La vraie question est de repérer tôt ce qui peut être stabilisé, de sécuriser les repas, de maintenir la mobilité et d’organiser un suivi pluridisciplinaire sérieux. C’est souvent là que se joue l’écart entre une maladie simplement subie et une maladie accompagnée avec méthode.