La fibrodysplasie ossifiante progressive est l’une des maladies génétiques les plus déroutantes du corps humain : elle transforme progressivement des tissus mous en os, là où aucune ossification ne devrait apparaître. Cet article explique ce qui se passe réellement dans l’organisme, pourquoi la mutation du gène ACVR1 suffit à tout dérégler, quels signes doivent alerter dès l’enfance et comment la prise en charge est organisée en France. L’objectif est simple : donner une lecture claire, utile et sans dramatisation inutile d’une maladie rare, mais très exigeante pour les patients et leurs proches.
Les points essentiels à garder en tête
- La FOP provoque une ossification hétérotopique, c’est-à-dire la formation d’os dans les muscles, tendons et ligaments.
- Le signe d’appel classique est souvent une déformation congénitale des gros orteils, déjà présente à la naissance.
- Des poussées inflammatoires douloureuses peuvent précéder l’apparition d’une nouvelle zone osseuse.
- La cause la mieux établie est une mutation gain de fonction du gène ACVR1, le plus souvent apparue de façon isolée.
- Le diagnostic repose surtout sur l’examen clinique et le test génétique, avec une règle majeure : éviter les gestes traumatiques tant que la maladie n’est pas exclue.
- En France, la prise en charge est surtout préventive, symptomatique et spécialisée, car il n’existe pas encore de traitement curatif de routine.
L’os se forme au mauvais endroit et verrouille le mouvement
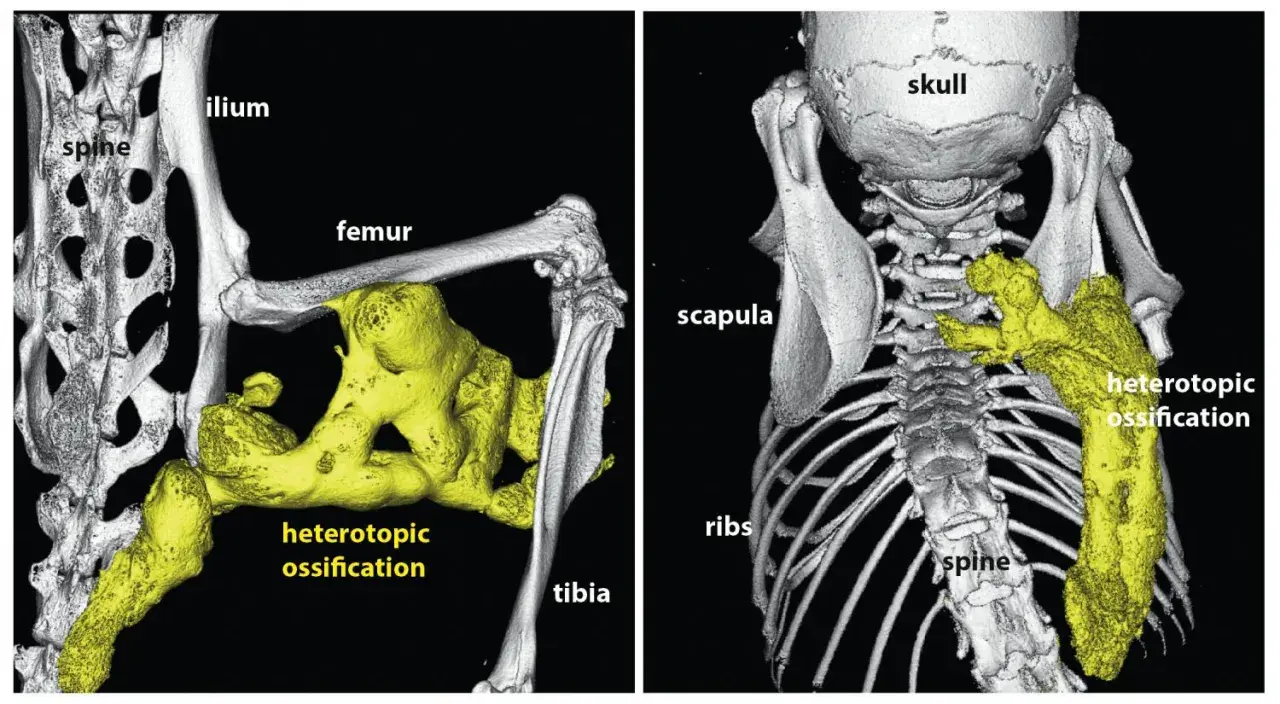
Je décris souvent cette maladie comme un bug profond du programme de réparation du corps. Normalement, lorsqu’un tissu est blessé, l’inflammation se calme puis la cicatrisation fait son travail. Dans la FOP, ce mécanisme déraille : le corps fabrique de l’os dans les tissus mous, ce qu’on appelle une ossification hétérotopique.
Le problème n’est pas seulement la présence d’un os “en trop”. C’est surtout son emplacement. Les muscles, les tendons, les ligaments et les fascias perdent peu à peu leur souplesse, puis leurs articulations deviennent difficiles à mobiliser. La maladie progresse souvent par étapes, avec une atteinte qui débute volontiers près du tronc, puis gagne les membres, la mâchoire et parfois la cage thoracique.
| Processus normal | Ce qui se passe dans la FOP | Conséquence concrète |
|---|---|---|
| Une blessure déclenche une inflammation brève puis une réparation localisée | Le signal de réparation s’emballe et oriente les cellules vers un programme osseux anormal | Des zones de tissu mou se transforment progressivement en os |
| L’inflammation est transitoire | Des poussées douloureuses, chaudes et gonflées peuvent revenir | Chaque poussée peut laisser derrière elle une nouvelle ossification |
| Les mouvements restent possibles après guérison | Les segments du corps se rigidifient au fil du temps | Perte de mobilité, gêne pour marcher, s’habiller, manger ou respirer |
La bonne façon de comprendre la maladie, c’est donc de voir l’os comme un tissu qui se construit au mauvais moment et au mauvais endroit. Et c’est précisément ce dérèglement qui amène à la question suivante : pourquoi le corps se met-il à fabriquer cet os parasite ?
Pourquoi une mutation du gène ACVR1 suffit à dérégler la réparation osseuse
La cause la mieux établie est un variant pathogène du gène ACVR1, aussi appelé ALK2. Ce gène code un récepteur impliqué dans la voie BMP, un circuit biologique qui participe normalement à la formation osseuse et au développement du squelette. Ici, la mutation agit comme un accélérateur bloqué : on parle de mutation gain de fonction, car le récepteur devient trop actif et envoie des signaux inadaptés.
Du point de vue génétique, la maladie suit un mode autosomique dominant : une seule copie altérée du gène suffit pour déclencher la maladie. Mais dans la pratique, la plupart des cas ne sont pas hérités d’un parent atteint. Ils apparaissent de façon sporadique, à la suite d’une mutation survenue “de novo”, c’est-à-dire de manière isolée chez un enfant sans antécédent familial clair.
- Si un parent est atteint, chaque enfant a un risque de transmission de 50 %.
- Si la mutation est de novo, le risque de récidive familiale est faible, mais pas nul, à cause de la possibilité d’une mosaïque germinale.
- Aucune prédisposition ethnique ou géographique nette n’a été identifiée, ce qui confirme le caractère ultra-rare mais universel de la maladie.
Je trouve ce point important, parce qu’il évite une idée fausse très répandue : l’absence d’antécédents familiaux ne doit jamais faire écarter la FOP trop vite. C’est justement ce qui explique tant de diagnostics tardifs, et cela nous mène aux signes précoces à ne pas banaliser.
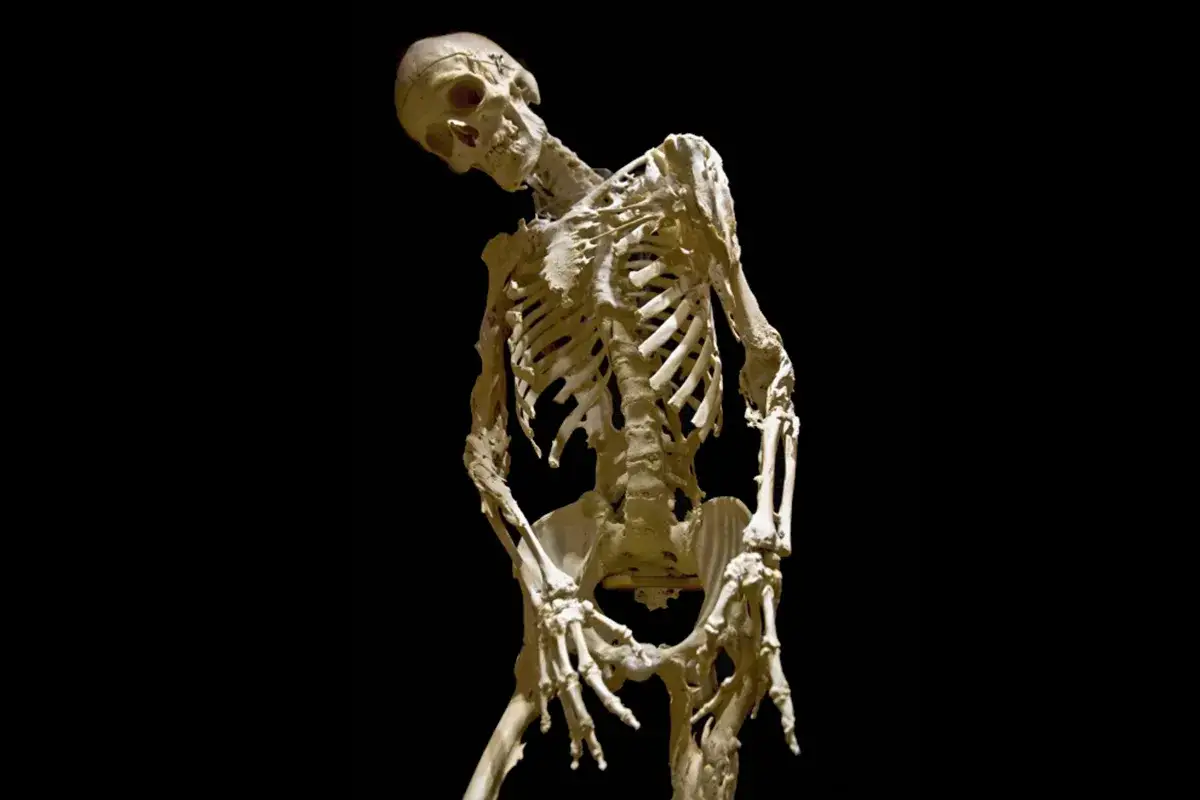
Les premiers signes à repérer dès l’enfance
Le premier indice le plus classique est une anomalie congénitale des pieds, le plus souvent des deux côtés. Le gros orteil peut être dévié, raccourci ou associé à un premier métatarsien anormal. Dit simplement, le pied “annonce” la maladie avant que l’os n’envahisse le reste du corps.
Ensuite viennent souvent les poussées inflammatoires : une zone devient douloureuse, chaude, gonflée, parfois rouge, puis se durcit progressivement. Cette évolution est trompeuse, car elle peut faire penser à une infection, à une tumeur bénigne ou à un traumatisme banal. Chez certains nourrissons, des nodules du cuir chevelu peuvent aussi apparaître très tôt et constituent parfois un premier signal discret.
| Signe | Ce que cela évoque | Pourquoi c’est important |
|---|---|---|
| Déformation congénitale des gros orteils | Indice très évocateur d’une FOP classique | Permet de penser à la maladie avant les premiers épisodes d’ossification |
| Gonflement douloureux, chaud, parfois rouge | Poussée inflammatoire, souvent appelée flare-up | Peut précéder une nouvelle zone d’os hétérotopique |
| Raideur du cou, du dos ou des épaules | Début d’atteinte axiale | Annonce une perte progressive d’amplitude articulaire |
| Difficulté à manger ou à parler | Atteinte de la mâchoire ou de la région cervico-faciale | Peut devenir rapidement handicapante au quotidien |
| Gêne respiratoire ou thorax qui se raidit | Atteinte de la cage thoracique | Doit alerter, car la respiration peut être compromise |
La difficulté, c’est que la maladie ne se présente pas toujours d’un seul bloc. Elle avance par petites étapes, parfois espacées, ce qui donne l’illusion d’un problème localisé. Or, c’est précisément cette lenteur trompeuse qui conduit souvent au retard diagnostique.
Comment le diagnostic doit être posé sans provoquer de dégâts
Le diagnostic de cette maladie doit être posé avec prudence. Quand la FOP est suspectée, il faut éviter les gestes traumatiques tant qu’elle n’est pas écartée : biopsie, injection intramusculaire, chirurgie non urgente, certains soins dentaires invasifs ou manipulations répétées peuvent déclencher ou aggraver une ossification.
En pratique, le médecin s’appuie d’abord sur l’examen clinique et sur l’histoire des symptômes. Les radiographies des pieds peuvent montrer des gros orteils caractéristiques, et le test génétique sur prise de sang permet de confirmer la présence d’un variant pathogène d’ACVR1. C’est un point central : dans une maladie aussi rare, le bon test au bon moment évite des erreurs coûteuses.Le retard diagnostique reste malheureusement fréquent. Dans les séries publiées, près de 90 % des patients reçoivent d’abord un autre diagnostic, et une grande partie subit des procédures inutiles qui peuvent laisser des séquelles durables. Pour être direct, c’est l’une des maladies où une mauvaise intuition initiale peut faire beaucoup de dégâts.
- À suspecter si les gros orteils sont anormaux dès la naissance et si des gonflements douloureux réapparaissent.
- À confirmer par un test génétique ciblé sur le sang.
- À éviter avant confirmation : biopsie, injection dans le muscle, chirurgie élective, gestes dentaires agressifs.
Le message clé est simple : quand le tableau clinique est évocateur, il vaut mieux protéger le patient tout de suite que “chercher à tout prix” par des gestes invasifs. Cette logique de prudence change ensuite complètement la manière de prendre en charge la maladie au quotidien.
Ce qui aide réellement au quotidien
Il n’existe pas, à ce jour, de traitement curatif de routine. La prise en charge repose donc sur trois axes très concrets : prévenir les traumatismes, traiter les poussées quand elles surviennent et préserver au maximum la fonction respiratoire, alimentaire et articulaire. En France, le suivi gagne à être coordonné par un centre expert des maladies osseuses constitutionnelles, car la FOP demande des réflexes médicaux très spécifiques.
Je insiste souvent sur un point : dans cette maladie, tout ce qui “force” le corps peut coûter plus cher qu’ailleurs. Cela vaut pour le sport, la kinésithérapie mal adaptée, certains soins dentaires ou des gestes médicaux improvisés.
| Situation | Ce qui est généralement utile | Ce qu’il vaut mieux éviter |
|---|---|---|
| Poussée inflammatoire | Anti-inflammatoires, parfois corticoïdes oraux selon la localisation et le contexte | Massage profond, reprise d’effort trop rapide, pression répétée sur la zone |
| Soins dentaires | Préparation avec une équipe habituée à la FOP, prévention bucco-dentaire précoce | Actes invasifs sans anticipation, anesthésie improvisée, ouverture forcée de la mâchoire |
| Mobilité | Aides techniques, hydrothérapie douce, adaptation du domicile, exercices respiratoires adaptés | Étirements passifs, sports de contact, fatigue musculaire intense |
| Risque de chute | Prévention à la maison, chaussures adaptées, protection de la tête chez l’enfant si besoin | Environnement encombré, activités à impact, chutes répétées |
Je retiens aussi une nuance utile pour les proches : la kinésithérapie n’est pas interdite, mais elle doit être pensée avec une extrême prudence. Le but n’est pas de “dérouiller” un membre par la force ; le but est d’entretenir ce qui peut l’être sans déclencher de traumatisme supplémentaire. C’est une différence capitale.
Pourquoi la vigilance précoce change la trajectoire de la maladie
La FOP reste une maladie grave et progressive. Les atteintes s’accumulent avec le temps, et les complications thoraciques et respiratoires comptent parmi les risques majeurs. C’est aussi pour cela que le diagnostic rapide est si important : il ne guérit pas la maladie, mais il permet souvent d’éviter des gestes iatrogènes, c’est-à-dire des dommages causés par les soins eux-mêmes.
Les recherches avancent, notamment autour de cibles moléculaires liées à ACVR1 et à l’activation anormale de la voie osseuse. C’est une vraie zone d’espoir, mais il faut rester rigoureux : en pratique, la prise en charge repose encore surtout sur la prévention, l’éducation, la surveillance et les traitements symptomatiques adaptés à chaque poussée.
Si je devais résumer la situation en une phrase, je dirais ceci : plus la FOP est reconnue tôt, plus on peut protéger le patient des erreurs qui accélèrent la perte de mobilité. Pour une maladie aussi rare, ce n’est pas un détail, c’est ce qui fait la différence entre un parcours chaotique et une prise en charge réellement maîtrisée.
Pour le lecteur, l’essentiel est donc de retenir les trois signaux d’alerte qui comptent vraiment : des orteils anormaux dès la naissance, des poussées inflammatoires douloureuses et une raideur qui progresse sans explication logique. Quand ce trio apparaît, la bonne réaction est de penser à la FOP vite, puis d’orienter vers un spécialiste sans multiplier les gestes agressifs.